Recent Highlights
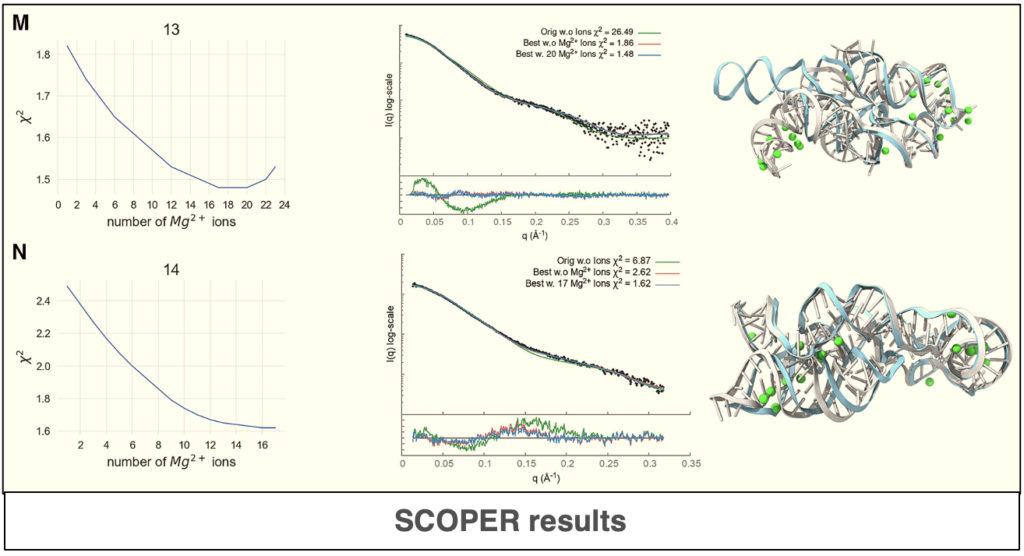
Predicting RNA structure and dynamics with deep learning and solution scattering
The use of deep-learning and statistical methods plays a significant role in the prediction of accurate structure and atomistics RNA models. In this paper, the authors describe new a deep-learning tool[…]
Read more
7th DNA Repair/Replication Structures and Cancer Conference : 24 Feb 2026 – 28 Feb 2026 in Playa Mujeres, Mexico
Integrative Structural Biology conference: The 7th DNA Replication/Repair Structures & Cancer Conference (7th DRRSC) will bring together scientists to exchange cutting-edge structural findings and stimulate new ideas and approaches to[…]
Read more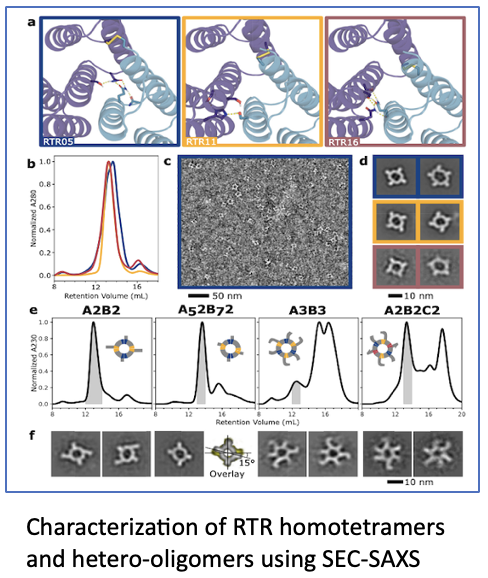
SEC-SAXS highlighted in Baker paper
In this Baker paper, published in Nature Communications in November 2024, the authors describe the design of pseudosymmetric protein hetero-oligomers. These proteins are versatile building blocks for creating functional materials[…]
Read more
Nobel Prize in Chemistry awarded to longtime SIBYLS user David Baker
David Baker, one of SIBYLS longest and most committed users, was awarded the Nobel Prize for Chemistry for his work in protein design. SAXS data collected from around 3,000 samples[…]
Read more