Recent Highlights
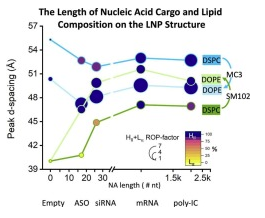
Exploring the impact of nucleotide length on lipid nanoparticle structure and properties
Using SAXS, supported by cryo-EM, SIBYLS scientists along with researchers at Genentech gained insights into the impact of nucleotide length on lipid nanoparticle structure and properties. LNPs serve as a[…]
Read more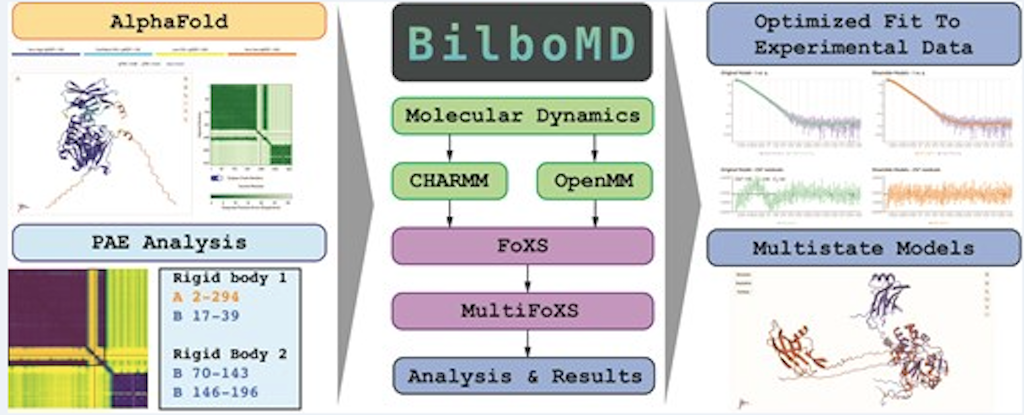
BilboMD: a web-accessible SAXS and AlphaFold-guided modeling pipeline
BilboMD is a web-based application developed and hosted by the SIBYLS beamline staff offering accessible pipelines for the analysis of SAXS data, including conformational sampling through molecular dynamics simulations, SAXS[…]
Read more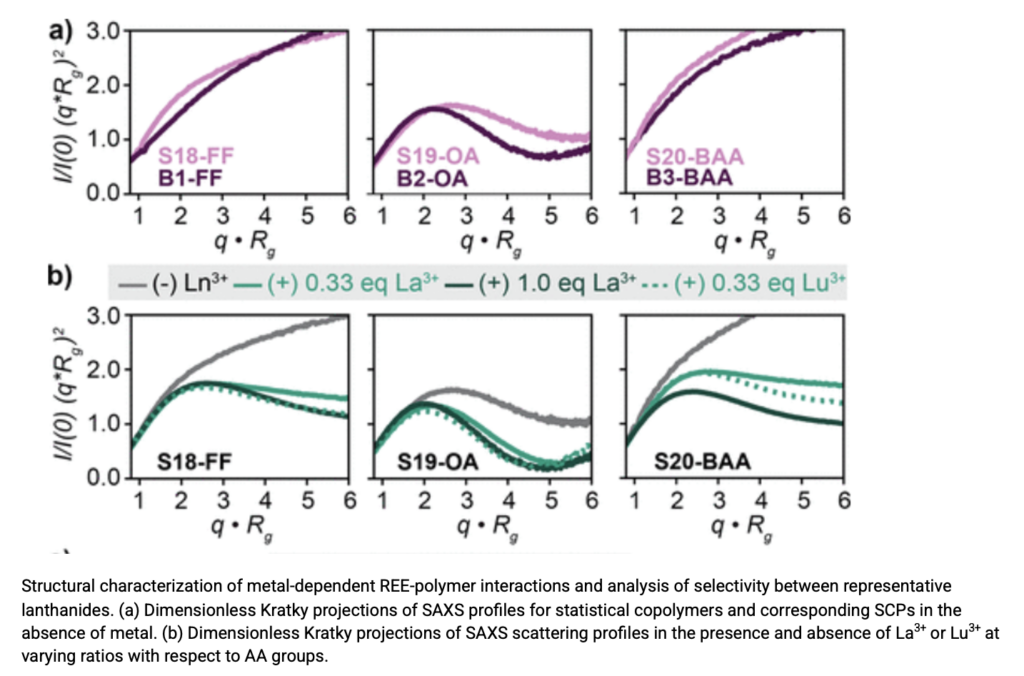
Structure–Function Relationships in Sequence-Controlled Copolymers for Rare Earth Element Chelation
In this publication led by Abigail Knight, an Assistant Professor of Chemistry at UNC-Chapel Hill and a SIBYLS SAXS user, the authors report on systematic research into amphiphilic polymer chelators,[…]
Read more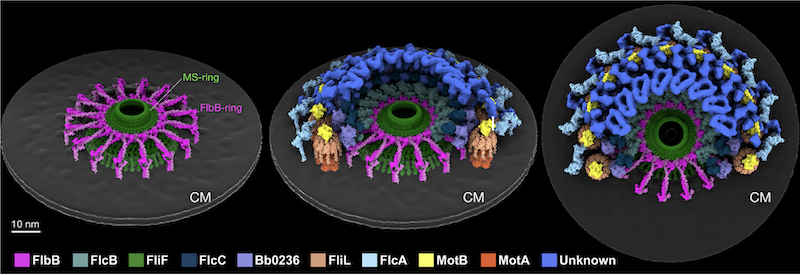
FlbB forms ring for assembly and motility
For this publication, the authors utilized SEC-SAXS data collected at SIBYLS beamline 12.3.1, along with SAXS model fitting, to confirm and further analyze the proteins of interest. Beamline scientist Michal[…]
Read more